
Clinical Care Guidelines
- Introduction Clinical Care Guidelines
- Chapter 1 The Fanconi Anemia DNA Repair Pathway
- Chapter 2 Diagnosis of Fanconi Anemia: Testing and Genetic Counseling
- Chapter 3 Clinical Care of Fanconi Anemia Hematologic Issues
- Chapter 4 Non-HNSCC Solid Tumors in Patients with Fanconi Anemia
- Chapter 5 Head and Neck Cancer in Patients with Fanconi Anemia
- Chapter 6 Oral Health Care for Patients with Fanconi Anemia
- Chapter 7 Gynecologic Care for Female Patients with Fanconi Anemia
- Chapter 8 Dermatologic Issues in Patients with Fanconi Anemia
- Chapter 9 Clinical Care of Fanconi Anemia Gastrointestinal Issues
- Chapter 10 Endocrine Disorders in Patients with Fanconi Anemia
- Chapter 11 Hearing and Ear Issues in Patients with Fanconi Anemia
- Chapter 12 Clinical Care of Hand and Arm Abnormalities in Fanconi Anemia
- Chapter 13 Brief Guide to Clinical Care for Patients with Fanconi Anemia
- Appendix A: Glossary and List of Abbreviations
- List of Contributors
Chapter 1
The Fanconi Anemia DNA Repair Pathway
Introduction
Discovery of the genes that cause Fanconi anemia (FA) and the role of FA proteins in regulating DNA repair have been active areas of research over the last 30 years.
In the last edition of the clinical care guidelines published by the Fanconi Anemia Research Fund in 2014, 16 FA genes had been discovered. Researchers have now identified 23 genes that, when mutated, cause FA, including FANCA [1], FANCB [2], FANCC [3], FANCD1/BRCA2 [4], FANCD2 [5], FANCE [6], FANCF [7], FANCG [8], FANCI [9,10,11], FANCJ/BRIP1 [12], FANCL [13], FANCM [14, 15, 16, 17], FANCN/PALB2 [18], FANCO/RAD51C [19, 20], FANCP/SLX4 [21,22], FANCQ/ERCC4 [23], FANCR/RAD51 [24, 25], FANCS/BRCA1 [26], FANCT/UBE2T [27, 28, 29], FANCU/XRCC2 [30], FANCV/REV7 [31], FANCW/RFWD3 [32], and FANCY/FAP100 [33].
The Fanconi Anemia DNA Repair Pathway
The Fanconi anemia (FA) proteins participate in a coordinated set of events that lead to the repair of interstrand crosslinks (ICLs) when the FA DNA repair pathway is activated during DNA replication (Figure 1, reviewed in [34, 35, 36]). The key event in the FA DNA repair pathway is the monoubiquitination of the FANCI and FANCD2 proteins (commonly referred to as the I-D2 or D2-I complex) [5, 9, 10, 11, 37, 38]. Monoubiquitination of the I-D2 complex depends on FANCL, an E3 ubiquitin ligase which works together with FANCT/UBE2T, an E2 ubiquitin-conjugating enzyme [27, 28, 29, 39]. FANCL is a component of a multi-subunit complex called the FA core complex. The core complex contains FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and the FA associated proteins (FAAP) FAAP20 and FAAP100 [40, 41, 42]. Deficiency of any of the core complex subunits leads to FA [1, 2, 3, 6, 7, 8, 13]. FANCM together with FAAP24 associates with the FA core complex, but its absence only partially reduces FANCI and FANCD2 ubiquitination [43, 44] and patients with biallelic mutations in FANCM have distinct presentation from the majority of FA patients (see Chapter 2) [16, 17, 45, 46]. FANCM is thought to participate in the activation of the FA pathway by localizing the core complex to chromatin and also through its role in activation of ataxia telangiectasia and Rad3-related (ATR), a DNA damage responsive kinase that phosphorylates multiple FA proteins [47]. FANCM also has been shown to be necessary for an early event during ICL repair, to promote skipping or “traverse” of the ICL lesion by the replication machinery [48, 49].
The FA core complex is necessary for the activation of its catalytic subunit, FANCL, and also for positioning its two substrates, FANCI and FANCD2, in proper orientation for monoubiquitination [50, 51]. The monoubiquitinated I-D2 complex stably localizes to lesions by encircling the DNA [52, 53]. Monoubiquitinated FANCI and FANCD2 are thought to facilitate the downstream repair events of DNA cutting and repair of the cut DNA [54, 55]. FANCQ/XPF is the essential nuclease that makes incisions around the lesion, a step called “unhooking” [23, 56]. XPF unhooking activity in this step is dependent on FANCP/SLX4, a protein scaffold for three nucleases [21, 22, 56, 57, 58, 59, 60]. Once the lesion is unhooked, a specialized translesion polymerase, Polζ, which includes FANCV/REV7 [31, 54], synthesizes DNA across the unhooked lesion creating a substrate for homologous recombination (HR) proteins. FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2, 11 FANCO/RAD51C, FANCR/RAD51, FANCS/BRCA1, FANCU/XRCC2 and FANCW/RFWD3 are proteins that participate in or regulate HR during ICL repair [4, 12, 18, 19, 24, 26, 30, 32, 61, 62, 63 64]. Some of the HR proteins, FANCR/RAD51 and FANCD1/BRCA2, also work at earlier steps of repair to protect the DNA at the ICL from inappropriate degradation by the DNA2 nuclease-Werner syndrome ATP-dependent helicase (WRN) complex [24, 64]. It is also important to note that the homologous recombination proteins indicated above participate in repair of DNA double-stranded breaks during DNA replication and are major tumor suppressors in the cell (reviewed in [65]). Once the ICL repair is completed, the pathway is turned off by a deubiquitinating enzyme, ubiquitin specific peptidase 1 (USP1) [66].
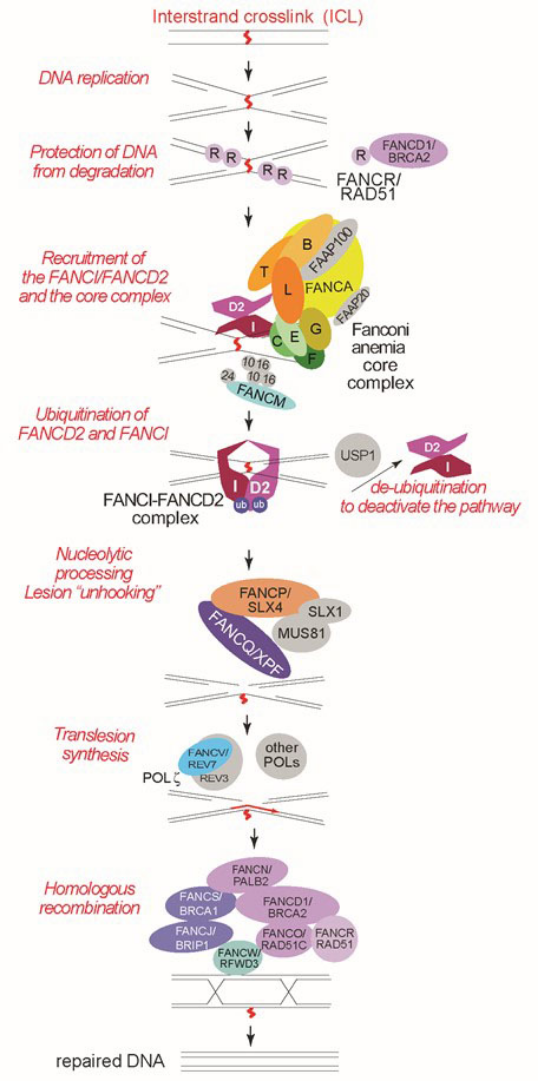
Figure 1. The role of Fanconi anemia proteins in the repair of DNA interstrand crosslinks (ICLs).
This simplified model highlights the activity of Fanconi anemia proteins. It is based on work from many laboratories as referenced in the text. For a more thorough review of the molecular pathway and additional proteins that participate in ICL repair, please see recent reviews [34-36]. For the latest study on the earliest steps of the pathway regulation during replication by the TRAIP protein, see reference [75] and for the studies on ICL traverse see references [48, 49, 76, 77]. See text for details.
In addition to participating in ICL repair, FA proteins are also active at other sites in the genome during DNA replication where they respond to replication stress, which is defined as any event that stalls the replication machinery. The FA proteins protect the newlyreplicated (nascent) DNA strands when replication is stalled [67, 68] (reviewed in [69]). They function at sites of under-replicated DNA, known as common fragile sites [70, 71, 72], and have been shown to play a role in the clearance of DNA:RNA hybrids, which form during transcription and are enhanced by replication and transcription machinery collisions [73, 74].
The Fanconi Anemia DNA Repair Pathway and Toxic Metabolites
The underlying pathophysiology of Fanconi anemia (FA) is best understood for the hematopoietic system, although more research in this area is needed. Hematopoietic stem cells (HSC) with a faulty FA pathway have increased levels of DNA damage and their attrition is an outcome of activation of the p53 pathway, which initiates apoptosis [78]. It also has been shown that driving quiescent/resting HSCs to enter the cell cycle by chronic bleeding or induction of type I interferon response can trigger bone marrow failure (BMF) in FA-deficient mice [79].These findings are consistent with the FA DNA repair pathway responding to problems encountered during DNA replication. These problems may occur at the sites of replication stress as defined above or may occur at the sites of DNA ICLs caused by metabolites that accumulate when stem cells divide or differentiate.
Endogenous metabolites, especially reactive aldehydes, including acetaldehyde and formaldehyde, have been strongly implicated in the pathogenesis of HSC failure and tumorigenesis in FA [80, 81, 82, 83]. Mice deficient in ALDH2, the enzyme which detoxifies acetaldehyde, develop bone marrow failure and leukemia [80, 81], which are common outcomes observed in patients with FA (see Chapter 3). Lack of the ADH5 enzyme, which participates in formaldehyde detoxification, leads to the development of bone marrow failure even faster in a mouse model of FA deficiency [82]. The mouse studies identifying genetic interaction between acetaldehyde and the FA DNA repair pathway have been corroborated in patients with FA. A dominant negative variant in ALDH2 (ALDH2*2, rs671 G>A) is common in East Asian peoples, including Han Chinese, Japanese, and Koreans [84]. Carriers of this variant have greatly diminished enzymatic activity and biallelic variants result in almost absent activity [85, 86]. Hira et al. studied a cohort of Japanese FA patients and showed that the patients who carried the ALDH2*2 variant displayed an increased number of congenital abnormalities, earlier onset of bone marrow failure, myelodysplastic syndrome, and leukemia [83]. Presence of the biallelic ALDH2*2 variants exacerbated the FA-associated phenotypes even more dramatically, leading to a severe disease presentation.
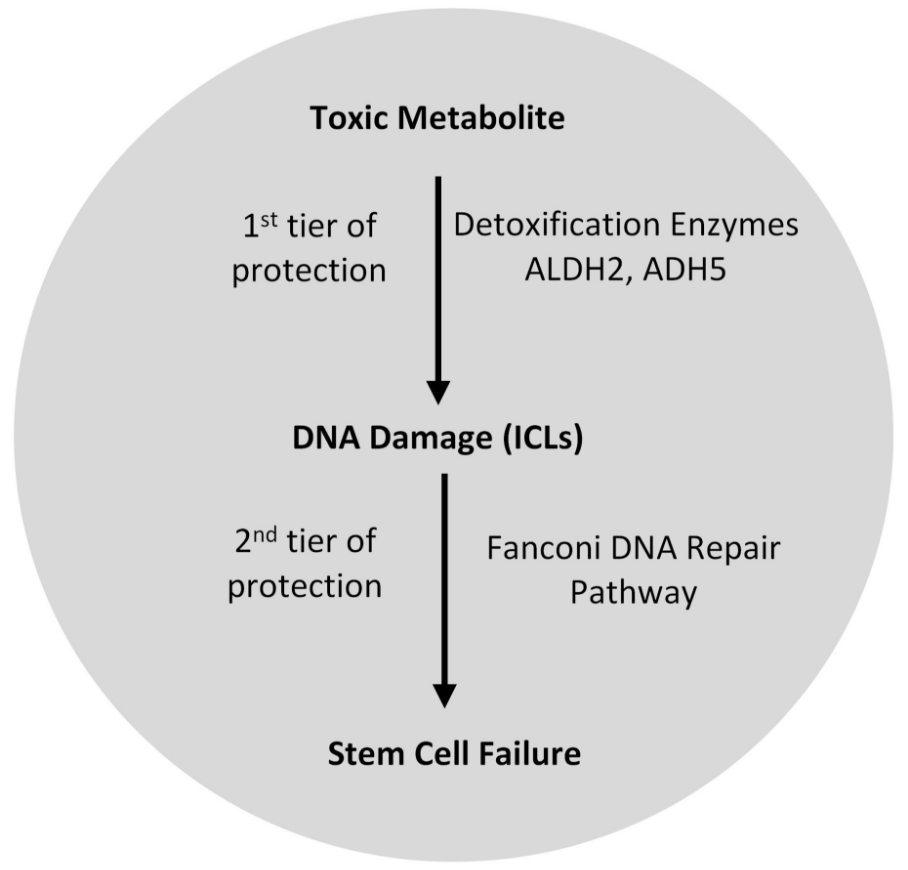
Figure 2. Two-tiered system for protection of hematopoietic stem cells [87].
The first tier, which includes ALDH2 and ADH5, detoxifies metabolites (acetaldehyde and formaldehyde, respectively) that have potential to damage DNA. The Fanconi anemia DNA repair pathway repairs the lesions made by metabolites that escape detoxification.
In light of the above studies, it is useful to think of a two-tier system to illustrate how mammalian cells protect themselves from endogenous toxic metabolites (Figure 2, reviewed in [87]). As toxic metabolites are produced by normal cellular metabolism or as a result of ingestion (for example alcohol), enzymes including aldehyde and alcohol dehydrogenases, detoxify the toxic metabolites into non-toxic molecules. Even with a fully functioning first tier of protection, some toxic metabolites escape detoxification and cause DNA lesions. The FA DNA repair pathway, the second tier of protection, is then needed to remove the resulting DNA lesions to prevent cell death or mutagenesis. In the scenario where more lesions result due to the deficiency of the first tier of protection of enzymatic detoxification, cells are even more reliant on an efficient DNA repair pathway due to a greater burden of damaging DNA lesions. Thus, deficiency of both tiers of protection leads to severe disease. This paradigm may be used outside of hematopoietic stem cells and for epithelial cells from which squamous cell carcinomas (SCC) develop. This would be an important association considering patients with FA have an increased risk of developing SCC (see Chapters 4, 5, 6, 7). However, the toxic metabolite implicated in generating DNA lesions likely will be dependent on tissue type. Understanding the sources of the endogenous DNA damage in FA cells will certainly contribute to understanding of the pathogenesis of the disease and may lead to development of new therapies.
Summary
Fanconi anemia (FA) is caused by faulty DNA repair of interstrand crosslinks (ICLs) in every cell in patients with the disease. Researchers have discovered 23 genes to date that cause FA and have established many key molecular mechanisms that regulate the FA DNA repair pathway. The absence of functional repair of ICLs can affect stem cells in particular, which results in many of the phenotypic manifestations associated with the disease. Bone marrow failure resulting from hematopoietic stem cells exposed to endogenous or exogenous toxic metabolites in the context of faulty FA DNA repair has been well characterized. However, more research is needed to develop a comprehensive understanding of how a faulty FA DNA repair pathway causes FA and squamous cell carcinoma and how mechanisms regulating the pathway can be exploited for novel treatments for patients with FA.
The Fanconi Cancer Foundation recognizes the following author contributions:
Agata Smogorzewska, MD, PhD
- Previous Chapter: Introduction
- Next Chapter: Chapter 2
References
- 1. Lo Ten Foe et al., Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet, 1996. 14(3): p. 320-323.
- 2. Meetei et al., X-linked inheritance of Fanconi anemia complementation group B. Nat Genet, 2004. 36(11): p. 1219-1224.
- 3. Strathdee et al., Cloning of cDNAs for Fanconi’s anaemia by functional complementation. Nature, 1992. 356(6372): p. 763-767.
- 4. Howlett et al., Biallelic inactivation of BRCA2 in Fanconi anemia. Science, 2002. 297(5581): p. 606-609.
- 5. Timmers et al., Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell, 2001. 7(2): p. 241-248.
- 6. de Winter et al., Isolation of a cDNA representing the Fanconi anemia complementation group E gene. Am J Hum Genet, 2000.
- 67(5): p. 1306-1308.
- 7. de Winter et al., The Fanconi anaemia gene FANCF encodes a novel protein with homology to ROM. Nat Genet, 2000. 24(1):
- p. 15- 16.
- 8. de Winter et al., The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet, 1998. 20(3): p. 281- 283.
- 9. Smogorzewska et al., Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell, 2007.
- 129(2): p. 289-301.
- 10. Sims et al., FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol, 2007. 14(6): p.
- 564-567.
- 11. Dorsman et al., Identification of the Fanconi anemia complementation group I gene, FANCI. Cell Oncology, 2007. 29(3): p. 211-218.
- 12. Levitus et al., The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet, 2005. 37(9):
- p. 934-935.
- 13. Meetei et al., A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet, 2003. 35(2): p. 165-70.
- 14. Meetei et al., A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M.
- Nat Genet, 2005. 37(9): p. 958-963.
- 15. Singh, T.R., et al., Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia
- complementation group M. Blood, 2009. 114(1): p. 174- 80.
- 16. Bogliolo, M., et al., Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet Med, 2018. 20(4):
- p. 458-463.
- 17. Catucci, I., et al., Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer,
- chemotherapy toxicity and may display chromosome fragility. Genet Med, 2018. 20(4): p. 452-457.
- 18. Reid, S., et al., Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet,
- 2007. 39(2): p. 162-4.
- 19. Vaz, F., et al., Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet, 2010. 42(5): p. 406-9.
- 20. Jacquinet, A., et al., Expanding the FANCO/RAD51C associated phenotype: Cleft lip and palate and lobar holoprosencephaly, two rare
- findings in Fanconi anemia. Eur J Med Genet, 2018. 61(5): p. 257-261.
- 21. Kim, Y., et al., Mutations of the SLX4 gene in Fanconi anemia. Nat Genet, 2011. 43(2): p. 142-6
- 22. Stoepker, C., et al., SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet,
- 2011. 43(2): p. 138-41.
- 23. Bogliolo, M., et al., Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet, 2013.
- 92(5): p. 800-6.
- 24. Wang, A.T., et al., A Dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of
- homologous recombination. Mol Cell, 2015. 59(3): p. 478-90.
- 25. Ameziane, N., et al., A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun, 2015.
- 6: p. 8829
- 26. Sawyer, S.L., et al., Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov, 2015. 5(2): p. 135-42.
- 27. Rickman, K.A., et al., Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T
- subtype of Fanconi anemia. Cell Rep, 2015. 12(1): p. 35-41
- 28. Hira, A., et al., Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet, 2015.
- 96(6): p. 1001-7.
- 29. Virts, E.L., et al., AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of
- Fanconi anemia. Hum Mol Genet, 2015. 24(18): p. 5093-108.
- 30. Park, J.Y., et al., Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi
- anaemia gene. J Med Genet, 2016. 53(10): p. 672-80.
- 31. Bluteau, D., et al., Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest, 2016. 126(9): p. 3580-4.
- 32. Knies, K., et al., Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest, 2017. 127(8): p. 3013-27.
- 33. Keuhl, B., et al., Biallelic Mutaitons in FAAP100 Predispose to Fanconi Anemia, in Fanconi Anemia Research Fund Scientific
- Symposium. 2018.
- 34. Kottemann, M.C. and A. Smogorzewska, Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 2013.
- 493(7432): p. 356-63.
- 35. Zhang, J. and J.C. Walter, Mechanism and regulation of incisions during DNA interstrand crosslink repair. DNA Repair (Amst), 2014.
- 19: p. 135-42.
- 36. Niraj, J., A. Farkkila, and A.D. D’Andrea, The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol, 2019. 3: p. 457-78.
- 37. Garcia-Higuera, I., et al., Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell, 2001. 7(2): p. 249-62.
- 38. Taniguchi, T., et al., S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood, 2002.
- 100(7): p. 2414-20.
- 39. Machida, Y.J., et al., UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell, 2006. 23(4): p.
- 589-96.
- 40. Ling, C., et al., FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J, 2007.
- 26(8): p. 2104-14.
- 41. Ali, A.M., et al., FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA
- DNA repair pathway. Blood, 2012. 119(14): p. 3285-94.
- 42. Kim, H., et al., Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol, 2012. 19(2): p. 164-70.
- 43. Bakker, S.T., et al., Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet, 2009.
- 18(18): p. 3484-95.
- 44. Ciccia, A., et al., Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell, 2007. 25(3): p.
- 331-43.
- 45. Kasak, L., et al., Bi-allelic Recessive Loss-of-Function Variants in FANCM Cause Non-obstructive Azoospermia. Am J Hum Genet,
- 2018. 103(2): p. 200-12.
- 46. Yin, H., et al., A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med, 2019. 21(1): p. 62-70.
- 47. Collis, S.J., et al., FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core
- complex. Mol Cell, 2008. 32(3): p. 313-24.
- 48. Huang, J., et al., The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell, 2013.
- 52(3): p. 434-46.
- 49. Rohleder, F., et al., FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res,
- 2016. 44(7): p. 3219-32.
- 50. Shakeel, S., et al., Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature, 2019. 575(7781): p. 234-7.
- 51. Wang, S., et al., Structure of the Fanconi Anemia Core-UBE2T complex poised to ubiquitinate bound FANCI-FANCD2,. bioRxiv, 2019.
- 854158.
- 52. Alcón P, et al., FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat Struct Mol Biol.
- 2020. 27(3): p. 240-8.
- 53. Wang, R., et al. DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature, 2020. 580, p. 278–82.
- 54. Räschle, M., et al., Mechanism of replication-coupled DNA interstrand crosslink repair. Cell, 2008. 134(6): p. 969-80.
- 55. Knipscheer, P., et al., The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science, 2009.
- 326(5960): p. 1698-701.
- 56. Klein Douwel, D., et al., XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol
- Cell, 2014. 54(3): p. 460-71.
- 57. Svendsen, J.M., et al., Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell, 2009.
- 138(1): p. 63-77.
- 58. Fekairi, S., et al., Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases.
- Cell, 2009. 138(1): p. 78-89.
- 59. Munoz, I.M., et al., Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell, 2009.
- 35(1): p. 116-27.
- 60. Kim, Y., et al., Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood, 2013. 121(1): p. 54-63.
- 61. Long, D.T., et al., Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science, 2011. 333(6038): p. 84-7.
- 62. Elia, A.E., et al., RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol Cell, 2015. 60(2): p. 280-
- 93.
- 63. Inano, S., et al., RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to
- Facilitate Homologous Recombination. Mol Cell, 2017. 66(5): p. 622-34.e8
- 64. Rickman KA, et al. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links.
- Genes Dev. 2020. 34(11-12): p. 832-46.
- 65. Chen, C.C., et al., Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer.
- Annu Rev Cancer Biol, 2018. 2: p. 313-36.
- 66. Nijman, S.M., et al., The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell, 2005. 17(3): p. 331-9.
- 67. Schlacher, K., et al., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by
- MRE11. Cell, 2011. 145(4): p. 529-42.
- 68. Schlacher, K., H. Wu, and M. Jasin, A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to
- RAD51-BRCA1/2. Cancer Cell, 2012. 22(1): p. 106-16.
- 69. Rickman, K. and A. Smogorzewska, Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol,
- 2019. 218(4): p. 1096-1107.
- 70. Chan, K.L., et al., Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol, 2009. 11(6): p. 753-
- 60.
- 71. Howlett, N.G., et al., The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of
- common fragile site stability. Hum Mol Genet, 2005. 14(5): p. 693-701.
- 72. Naim, V. and F. Rosselli, The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome
- abnormalities. Nat Cell Biol, 2009. 11(6): p. 761-8.
- 73. Garcia-Rubio, M.L., et al., The Fanconi Anemia Pathway Protects Genome Integrity from Rloops. PLoS Genet, 2015. 11(11): p.
- e1005674.
- 74. Schwab, R.A., et al., The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell,
- 2015. 60(3): p. 351-61.
- 75. Wu, R.A., et al., TRAIP is a master regulator of DNA interstrand crosslink repair. Nature, 2019. 567(7747): p. 267-72.
- 76. Huang, J., et al., Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep,
- 2019. 27(6): p. 1794-1808 e5
- 77. Ling, C., et al., Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and
- traverse of DNA interstrand crosslinks. Cell Discov, 2016. 2: p. 16047.
- 78. Ceccaldi, R., et al., Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs
- hematopoietic stem and progenitor cells. Cell Stem Cell, 2012. 11(1): p. 36-49.
- 79. Walter, D., et al., Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature, 2015. 520(7548):
- p. 549-52.
- 80. Langevin, F., et al., Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature, 2011. 475(7354): p. 53
- 81. Garaycoechea, J.I., et al., Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature,
- 2012. 489(7417): p. 571-5
- 82. Pontel, L.B., et al., Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell, 2015.
- 60(1): p. 177-88.
- 83. Hira, A., et al., Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients.
- Blood, 2013. 122(18): p. 3206-9.
- 84. Yoshida, A., I.Y. Huang, and M. Ikawa, Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in
- Orientals. Proc Natl Acad Sci U S A, 1984. 81(1): p. 258-61.
- 85. Crabb, D.W., et al., Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH2(2) allele is
- dominant. J Clin Invest, 1989. 83(1): p. 314-6
- 86. Lai, C.L., et al., Dominance of the inactive Asian variant over activity and protein contents of mitochondrial aldehyde dehydrogenase
- 2 in human liver. Alcohol Clin Exp Res, 2014. 38(1): p. 44-50.
- 87. Garaycoechea, J.I. and K.J. Patel, Why does the bone marrow fail in Fanconi anemia? Blood, 2014. 123(1): p. 26-34.